


Direkte RNA-Sequenzierung

Nanostring ncounter von Nanostring (Quelle: © Nanostring).
Der Genotyp ist die genetische Ausstattung eines Organismus, der Phänotyp die Ausprägung seiner Merkmale. Dazwischen steht das Transkriptom, umgeben von zahlreichen RNA-Spezies. Hochdurchsatz-RNA-Sequenzierungstechnologien erlauben tiefe Einblicke in die Transkription und eröffnen neue Horizonte.
In den letzten 10 Jahren untersuchten Wissenschaftler die dynamischen Zustände zahlreicher Genome vom einfachen Modellorganismus bis zum komplexen, menschlichen Genom. Sie erfassten sowohl Modifikationen auf DNA-Ebene, als auch quantitative und qualitative Veränderungen der RNA. Heute haben wir ein neues Verständnis für die Komplexität eines Transkriptoms, das umgeben ist von einer Vielzahl bisher unbekannter kodierender und nicht-kodierender RNA Spezies, besonders den small RNAs (sRNAs) wie microRNAs, den promotorassoziierten RNAs und den erst kürzlich entdeckten antisense 3`-termini-assoziierten RNAs, um nur einige zu nennen.
Microarrays als Standardtechnologie
Anfänglich wurden Transkriptomstudien hauptsächlich mit DNA-Microarrays durchgeführt. Seit ihrer Erfindung im Jahr 1991 sind DNA-Microarrays oder Genchips zwar heute immer noch die am häufigsten genutzte Technik zur Expressionsanalyse von Genen, sie werden aber durch die immer kostengünstiger werdenden Hochdurchsatz-Next-Generation DNA-Sequenzierungsmöglichkeiten (NGS) ergänzt und in zahlreichen Anwendungen möglicherweise auch bald ersetzt werden.
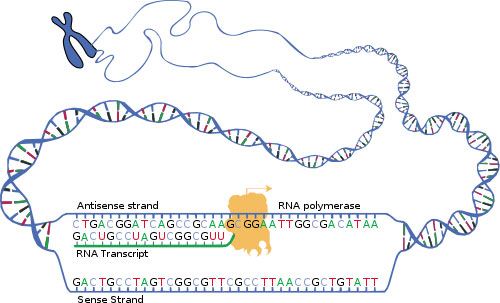
Transkriptionsschema.
Bildquelle: © National Human Genome Research Institute/wikimedia.org; public domain
Wegen des hohen parallelen Probendurchsatzes, der vergleichsweise geringen Probenmenge und der guten Automatisierbarkeit hatte sich die Microarraytechnik rasch als wichtiges Untersuchungswerkzeug in der Forschung für die Bereiche Pharmazie, Medizin, Biochemie, Genetik und Molekularbiologie durchgesetzt. Überzeugt hat die Microarraytechnologie außerdem aufgrund ihres geringen Arbeitsaufwandes und der überschaubaren Kosten für derartige Experimente. Doch nur günstig und schnell zu sein reicht bei den Anforderungen an biologische Untersuchungsmethoden nicht aus: Für zuverlässige Ergebnisse brauchen Wissenschaftler auch verlässliche, reproduzierbare und untereinander vergleichbare Untersuchungsergebnisse - und in diesen Punkten zeigen Microarrays Schwächen. Es gibt viele unterschiedliche Microarray-Technologien mit jeweils wiederum einer Vielzahl von Parametern. Dies führt dazu, dass die durch Microarrays gewonnenen Ergebnisse selten quantitativ vergleichbar und oft sogar bei ein und demselben Experiment schwer reproduzierbar sind. Bei Arbeiten mit selbst bedruckten DNA-Microarrays, etwa wenn es keine kommerziellen Arrays gibt, fällt die Reproduzierbarkeit der Ergebnisse noch schlechter aus. Oft fällt es Wissenschaftlern außerdem schwer, den erheblichen Datenmengen, die von einem Microarray erzeugt werden, eine biochemische Funktion zuzuordnen.
NGS-Methoden auf dem Vormarsch
Das Aufkommen der NGS-Geräte erlaubte RNA-Analysen durch cDNA-Sequenzierung im großen Stil. Die NGS-basierten Methoden zur RNA-Sequenzanalyse haben die Transkriptomanalyse in Pro- und Eukaryonten beträchtlich verbessert. So wurden beispielsweise in der Kartierung von Transkriptionsstarts, bei strangspezifischen Messungen, bei der Ermittlung von Genzusammenschlüssen, bei der Charakterisierung von kleinen RNAs und beim Aufdecken von alternativen Spleißingevents erhebliche Fortschritte seit der Einführung dieser neuen Technologien gemacht. NGS-Geräte zur RNA-Sequenzierung sind momentan von vier kommerziellen Herstellen erhältlich: Illumina, Roche 454, HelicosBiosciences und Life Technologies. Die Hersteller arbeiten ständig daran, die wichtigsten Sequenzierungsparameter wie Durchsatz, Leselänge, Fehlerquote und die Möglichkeit zur „paired end“ Analyse zu optimieren, um eine möglichst schnelle und fehlerarme Sequenzierung zu einem möglichst geringen Preis anbieten zu können. Neue Technologien von anderen Herstellern befinden sich außerdem in Entwicklung (Metzker, M. L., 2010).

Genchips - die heute noch am häufigsten genutzte Technik zur Expressionsanalyse von Genen.
Bildquelle: © Affymetrix / www.affymetrix.com
Wie funktioniert eine Standard-RNA-Sequenzierung?
Die meisten RNA-Sequenzierungsmethoden beruhen auf der cDNA Synthese, gefolgt von einer Reihe von Manipulierungsschritten, die je nach verwendetem System variieren. Sie basieren auf dem Prinzip der Pyrosequenzierung.
Probleme und Grenzen der RNA-Sequenzierung
Je nach Anwendung kann es bei der RNA-Sequenzierung zu unterschiedlichen Schwierigkeiten kommen.
- Verlust der Stranginformation
Für einen Standard-RNA-Sequenzierungsansatz benötigt man doppelsträngige cDNA als Ausgangsmaterial, wodurch die RNA-Strang-Information verloren geht. Dies ist vor allem bei strangspezifischen RNA-Sequenzierungen zur Charakterisierung von sog. Antisense- Transkriptionsevents von Bedeutung, die einst für biologisches oder technisches Rauschen gehalten wurden. Heute weiß man, dass Antisense-Transkripte eine Funktion haben und sowohl im physiologischen als auch im pathologischen Zustand eine Rolle spielen.
- Fehleranfällige reverse Transkriptase
Das Umschreiben der RNA in cDNA wird von einer RNA-abhängigen DNA-Polymerase (reverse Transkriptase) übernommen, die ihre Arbeit nicht immer sehr sorgfältig ausführt. Beim Umschreiben der RNA in den ersten Strang der cDNA verwechselt die reverse Transkriptase manchmal den „sense“ mit dem „antisense“ Strang. Das führt zu unechten cDNA Zweitsträngen, die eine Antisense-Transkriptanalyse ausschließen. Im Gegensatz zu anderen Transkriptasen besitzt die reverse Transkriptase außerdem keine Korrekturlesefunktion, was ihre Arbeit fehleranfällig macht. Die Effektivität der unterschiedlichen reversen Transkriptasen beim Umschreiben von RNA in cDNA schwankt außerdem abhängig von den Bedingungen im Experiment.
- Andere Störfaktoren
An anderen Störfaktoren trägt die reverse Transkriptase jedoch keine Schuld. So kann die während der reversen Transkription (dem Umschreiben von RNA in cDNA) entstehende cDNA von der Matrizen-RNA ab-dissoziieren und sich an einen RNA-Abschnitt mit einer ähnlichen Sequenz wie der Ausgangssequenz anlagern. Dies erzeugt künstliche, chimäre cDNA. Dieser sog. Matrizenwechsel führt beispielsweise zu Problemen bei der Identifizierung von Exon-Intron-Grenzen und echten chimären Transkripten.
Bilden sich sekundäre RNA-Strukturen, kann die cDNA-Synthese sogar Primer-unabhängig vonstatten gehen. Dann entsteht zufällig generierte cDNA, die die Messergebnisse verfälscht.
- Anreichern von „Abfall“
RNA-Sequenzierungen benötigen oft einen poly(A) mRNA-Anreicherungsschritt. Die Polyadenylierung (bzw. poly(A) bzw. Tailing) von Transkripten, also das Anhängen mehrerer Adenine an den mRNA-Strang, findet auch bei Degradierungsprozessen statt. Die nachfolgenden Anreicherungsschritte reichern dementsprechend auch unerwünschte RNA-Degradierungsprodukte an und verfälschen so die Messergebnisse.
- Kleine RNAs fallen durchs Raster
Nicht nur das die Nachlässigkeit der reversen Transkriptase und das Umschreiben von RNA in cDNA können zu Fehlern bei der RNA-Sequenzierung führen. Auch bei der Untersuchung von kleinen RNAs (engl. smallRNAs oder sRNAs) gibt es noch Verbesserungspotential: Untersuchungen zeigten, dass NGS-basierte RNA-Sequenzierungs-ansätze zwar zur differentiellen Expressionsanalayse von smallRNAs herangezogen werden können, dass sie aber nicht zu absolut quantitativen Messungen in der Lage sind.

BioMark-HD System von Fluidigm.
Bildquelle: © Fluidigm
Die Lösung: Direkte RNA-Sequenzierung
Je mehr Manipulationsschritte die zu sequenzierende RNA durchlaufen muss, desto fehleranfälliger ist die Sequenziermethode. Daher suchen NGS-Gerätehersteller nach neuartigen Verfahren, mit denen RNA-Moleküle direkt, ohne vorheriges Umschreiben in cDNA, in ihrer Sequenz erfasst werden können. Die Amplifikation von RNA-Molekülen - ohne die vorherige Transkription in cDNA - ist bisher noch nicht möglich. RNA-abhängige RNA-Polymerasen existieren zwar, in welcher Weise sie jedoch bei den NGS-Methoden zum Einsatz kommen könnten, ist bisher noch nicht erforscht.
- Die Pioniere
Der erste direkte parallele Sequenzierungsansatz von RNA wurde im Jahr 2009 mit der Einzel-Molekül-Sequenzierungsplattform von Helicos durchgeführt (vgl. Ozsolak et al. (2009)). Helicos verwendet dabei mit Poly(dT)-bestückte Träger, an die einige Femtomol 3`-polyadenylierte RNA-Moleküle binden. Die Sequenzierung jedes einzelnen Moleküls erfolgt während der Synthese des Gegenstrangs. Mit diesem Ansatz kann Poly(A) -RNA aus totaler RNA und aus Zelllysaten isoliert und sequenziert werden. Die Sequenzinformation beginnt direkt nach der Poly(A) -Sequenz. Daher eignet sich die Methode zum Genexpressionsprofiling und zur genomweiten und quantitativen Kartierung von Polyadenylierungssequenzen. RNAs ohne natürliche Polyadenylierungssequenz können in vitro polyadenyliert und anschließend sequenziert werden. Auch wenn diese Methode frei von cDNA Artefakten ist, bleibt dennoch das Risiko bestehen, Degradationsprodukte während der Selektion polyadenylierter RNAs zu sequenzieren.
- Auch für sehr geringe RNA-Ausgangsmengen geeignet
Das direkte Sequenzieren von RNA hat aber noch weitere Vorteile: Die Probenvorbereitung ist einfach und erfordert nur äußerst geringe Ausgangsmengen an RNA. Je nach Anwendung reichen Femto- oder sogar Attomol aus. Das macht die Methode hauptsächlich für Fragestellungen interessant, bei denen nur sehr wenig Probenmaterial zur Verfügung steht. Vor allem in der Forensik, der Stammzellbiologie, der Metagenomforschung und der Pflanzenbiologie sind solche Untersuchungen von besonderer Bedeutung. Auch in der Krebsforschung und im Bereich der personalisierten Medizin könnte die direkte RNA-Sequenzierung zu spürbaren Verbesserungen für die Patienten führen, da Patientenproben naturgemäß in ihrer Quantität begrenzt sind.
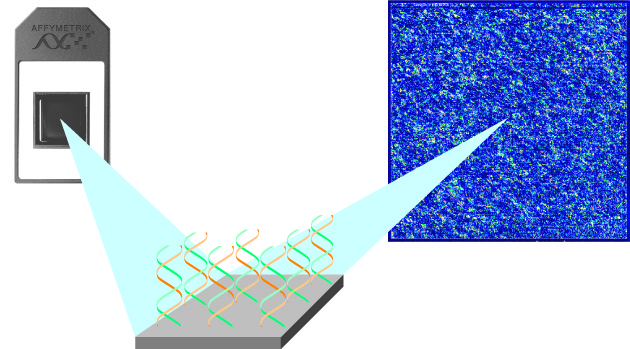
Schema der Genchiptechnologie.
Bildquelle: © Affymetrix / www.affymetrix.com
- Verschiedene neue Sequenzierungsansätze
Untersuchungsmethoden zum Profiling von RNA mit geringen Ausgangsmengen, die auf Sequenzierung basieren, sind relativ neu. Im Jahr 2009 berichtete eine britische Arbeitsgruppe in Zusammenarbeit mit Applied Biosystems von der gesamten mRNA-Sequenzierung eines einzelnen Oozyten (Eizelle). Die Reproduzierbarkeit der Methode erscheint allerdings noch verbesserungswürdig. (Tang, F. et al. (2009)).
In letzter Zeit etablieren sich immer mehr Methoden, mit denen Wissenschaftler verlässliche Transkriptomprofile von geringem RNA-Ausgangsmaterial erstellen können. Diese Methoden basieren entweder auf Sequenzierung oder auf Hybridisierung.
Auf Sequenzierung basiert beispielsweise die NanoCAGE (cap analysis of gene expression) Technologie (Referenz 12). Mit ihr können Transkriptionsstartsequenzen von 10 Nanogramm totaler RNA Ausgangsmenge durch verschiedene Amplifikationsschritte kartiert werden. (Plessy, C. et al. (2010)).
Ein anderer Ansatz kommt ohne Amplifikation aus. Dabei werden First-Strand cDNA Produkte von nur ungefähr 500 Picogramm RNA sequenziert; das Primen findet in Lösung mit Oligo-dTs oder zufälligen Hexameren statt (Lipson, D. et al. (2009)).
Mit einer weiteren Methode können reproduzierbare Genexpressionsprofile von einer Ausgangsmenge von etwa 1000 Zellen erstellt werden. Über immobilisierte poly(dT)-Primer wird poly(A) mRNA von zellulären Lysaten isoliert. Im Anschluss erfolgen cDNA-Synthese und Sequenzierung auf der Oberfläche. Durch das Anheften an eine Oberfläche wird vermieden, dass RNA während der RNA-Isolierungsschritte verloren geht, was bei geringen Ausgangsmengen von besonderer Bedeutung ist (Ozsolak, F. et al. (2010)).
Auf dem Prinzip der Hybridisierung basiert das NanoString nCounter System, das ohne cDNA-Synthese auskommt. Target-spezifische Sonden hybridisieren mit den RNA-Proben. Die Sonden-RNA-Duplexe werden auf einer Oberfläche immobilisiert und einzelne Moleküle zur Identifizierung optisch erfasst. Im Prinzip kann dieses System 16.384 Transkripte gleichzeitig detektieren. Dafür benötigt es etwa 100 Nanogramm RNA oder 2.000 bis 5.000 Zellen.
Fluidigm hat eine Mikroströmungsplattform entwickelt, die mittels quantitativer Real-Time PCR das Genexpressionsprofil einzelner Zellen erstellen kann. Der Ansatz ist vor allem bei der Bestimmung von Expressionsleveln bei einer Auswahl von Transkripten von Interesse.
Keines der beschriebenen Verfahren ist gänzlich ausgereift und keines kann dem Anspruch der Erstellung eines absolut zuverlässigen, genom-weiten und tiefgründigen Transkriptomprofil bei sehr kleinen Ausgangsmengen an RNA gänzlich gerecht werden. So arbeiten beispielsweise sowohl die Fluidigm- als auch die NanoString-Technologie nur mit einer ausgewählten Gruppe von Transkripten und eignen sich nicht zu Vergleichsanalysen. Die neuen Technologien sind zwar alle noch nicht perfekt, bieten aber eine solide Grundlage für zukünftige Verbesserungen je nach gewünschter Anwendung.

Dynamic Array von Fluidigm.
Bildquelle: © Fluidigm
Was bringen die neuen Technologien?
Die Sequenziertechnologie hat in den letzten 20 Jahren wahre Quantensprünge erlebt. Mitte der 90er Jahre wurden die klassischen DNA-Sequenziermethoden nach Sanger bzw. Maxam und Gilbert durch die Pyrosequenzierung zunächst ergänzt und heute praktisch abgelöst. Die Pyrosequenzierung bildete die Grundlage für die NGS-Technologien. Heute sind Wissenschaftler mit leistungsfähigen, aber immer noch verbesserungswürdigen Analysewerkzeugen zur Charakterisierung und Quantifizierung von Transkriptomen ausgestattet. Die kürzlich entwickelten Technologien versprechen zumindest eine teilweise Verbesserung der aktuellen Probleme, die mit der Sequenzierung von RNA-Molekülen einhergehen. Mit den zu erwartenden Verbesserungen in den nächsten Jahren werden Wissenschaftler in der Lage sein, komplette Transkripte - vom einfachen Einzeller-Genom bis hin zum komplexen Säugetier-Genom - in gesunden und in kranken Geweben zu katalogisieren. Auch wenn nur geringe RNA-Mengen zur Verfügung stehen, können komplexe biologische Netzwerke in einer Vielzahl biologischer Spezies in Zukunft definiert werden. Darauf aufbauend können Wissenschaftler virtuelle RNA-Netzwerk-Modelle von Zellen und Geweben erstellen, um die Stoffwechselwege zu verstehen, die unter den jeweils unterschiedlichen physiologischen Konditionen aktiv sind.
RNA-Bestimmungen können auch für die klinische Diagnose (z.B. bei der Analyse extrazellulärer Nukleinsäuren, fetaler RNA oder zirkulierender Tumorzellen) relevant sein. Tumorzellen können mit den neuen Technologien evtl. früher entdeckt und im Zuge der personalisierten Medizin individuell behandelt werden.
Die neuen RNA-Sequenzierungstechnologien fügen die Puzzelteile der Genominformation zusammen, sodass unser Verständnis für grundlegende biologische Fragen wie der Differenzierung von Zellen oder der biologischen Diversität nach und nach vervollständigt werden wird.
Quellen:
- Metzker, M. L. (2010): Sequencing technologies – the next generation. Nature Rev. Genet. 11, 31-46. (abstract).
- Tang, F. et al. (2009): mRNA-seq whole transcriptome analysis of a single cell. Nature Methods 6, 377-382. (abstract).
- Plessy, C. et al. (2010): Linking promotors to functional transcripts in small samples with nanoCAGE and CAGEscan. Nature Methods 7, 528-534. (abstract).
- Lipson, D. et al. (2009): Quantification of the yeast transcriptome by single-molecule sequencing. Nature Biotechnol. 27, 652-658. (abstract).
- Ozsolak, F. et al. (2010): Amplification-free digital gene expression profiling from minute cell quantities. Nature Methods 7, 619-621. (abstract).
Anregungen zum Weiterlesen:
- DNA-Sequenzierungsmethoden - Motoren der Genomforschung
- Ultra-Hochdurchsatz-Sequenziermethoden im Überblick
- Klassische Sequenzierungsmethoden im Überblick
- Interview mit Dr. Stangier (GATC Biotech AG) zu „Maßgeschneiderte DNA-Sequenzierung“
