


Dem Genom der Gerste auf der Spur

Gerstenfeld im Stadium der Milchreife. (Quelle: © IPK Gatersleben)
Das Gerstegenom ist groß und komplex. Seine Sequenzierung wird auch Informationen über das nah verwandte Weizengenom liefern. Wissenschaftler entwickelten nun eine erfolgreiche Strategie, wie das Genom der Gerste entschlüsselt werden kann.
Die wachsende Weltbevölkerung und der Klimawandel stellen die Menschheit vor neue Herausforderungen in der Erzeugung von Nahrungsmitteln. Die Genomsequenz einer Pflanze ist dabei der zentrale Ausgangspunkt für moderne Züchtungsmethoden, mit denen das volle genetische Potential der Pflanze ausgeschöpft werden kann. Nur wenn Wissenschaftler den genetischen Code einer Pflanze kennen, können sie wichtige Fragen der Nutzpflanzenbiologie beantworten und die molekularen Mechanismen verstehen, die für die Ausprägung komplexer Eigenschaften verantwortlich sind.

Gerstenkörner.
Bildquelle: © IPK Gatersleben
Bisher liegen nur die Genome von Reis (Oryza sativa) und der Modellpflanze Arabidopsis thaliana vollständig und in hoher Sequenzqualität vor. Die Größe und Komplexität der Getreidegenome erschwerten bisher deren Sequenzierung. Einer internationalen Forschungsgemeinschaft unter der Leitung von Dr. Nils Stein, Arbeitsgruppenleiter am vom Leibniz-Institut für Pflanzengenetik und Kulturpflanzenforschung (IPK) in Gatersleben, gelang nach zweijähriger Arbeit erstmalig der genomweite Einblick in die ökonomisch und wissenschaftlich wichtige Getreideart Gerste. Selbst wenn sie mit derzeitigen Methoden noch keine Referenzsequenz erstellen konnten, entwickelten die Wissenschaftler nun eine ausbaufähige Übergangslösung, die als Grundlage für die vollständige Genomsequenzierung von Gerste und Weizen dienen wird.
Warum gerade Gerste?
Gerste ist weitaus mehr als der Rohstoff für Bier und Viehfutter. Die Triticeae Spezies Weizen (Triticum aestivum) und Gerste (Hordeum vulgare) nehmen laut Daten der FAO (Food and Agricultural Organisation of the USA) gemessen an ihrer Anbaufläche Platz eins und fünf in der weltweiten Nahrungsmittelproduktion ein (FAOSTAT, 2007). Dass es von diesen beiden bedeutenden Getreidearten bisher noch keine vollständige Genomsequenz von brauchbarer Qualität gibt, liegt vor allem an deren Größe und Komplexität.
Das Weizengenom umfasst etwa 17 Gigabasenpaare (Gb), das der Gerste 5,1 Gb. „Das Gerstengenom ist etwa eineinhalb mal so groß wie das menschliche Genom und entspricht ungefähr 12 mal dem Reisgenom, dessen Sequenzierungskosten auf über 100 Millionen US-Dollar geschätzt wurden“, verdeutlicht Dr. Nils Stein die Größenverhältnisse. Weizen verfügt über ein hexaploides Genom (2n=6x=42), das aus der Verschmelzung dreier nah verwandter Genome entstanden ist. Das Genom der Gerste ist diploid (2n=2x=14). „Weizen und Gerste sind sehr nah verwandte Getreidearten mit vielen ähnlichen Eigenschaften und man weiß aus vergleichenden Genomkartierungen und aus zytogenetischen Untersuchungen, dass die Anordnung der Gene in Weizen und Gerste sehr ähnlich ist“, so Dr. Stein. Eine ähnliche Abfolge eines Großteils der Gene fanden Forscher auch beim Vergleich von Gerste und Weizen mit anderen Gräsergenomen, wie beispielsweise dem des Reis, der Hirse (Sorghum bicolor) oder des Wildgrases Zwenke (Brachypodium distachyon).

Versuchsfeld auf dem Gelände des Leibniz-Institut für Pflanzengenetik und Kulturpflanzenforschung (IPK).
Bildquelle: © IPK Gatersleben
Wissenschaftler bezeichnen die gemeinsame Reihenfolge von Genen oder Gensegmenten auf verschiedenen chromosomalen Abschnitten, wie sie beim Vergleich von Weizen und Gerste findet, als Kollinearität oder als konservierte Syntenie. Zwischen Gerste und Weizen sind viele Merkmale stark konserviert. Dazu gehören beispielsweise die Blühkontrolle, die Regulation von Winter- und Sommerwuchsform (Unterscheidung Winter-/Sommergetreide) oder die Wuchshöhe. „Die Urformen unserer heutigen Getreide Gerste und Weizen stammen aus derselben Region, dem sog. Gebiet des goldenen Halbmondes – der Südosttürkei, dem Iran, Nord-Syrien und Israel“, erklärt Dr. Stein. Ihre Entwicklungswege trennten sich erst vor etwa zwölf bis 13 Millionen Jahren, die von Reis und Gerste/Weizen bereits vor 50 bis 70 Millionen Jahren. Daher wird die Entschlüsselung des Gerstengenoms auch zum besseren Verständnis des weitaus komplexeren Weizengenoms beitragen.
Warum sich Forscher nicht gleich an den Weizen wagten, erklärt Dr. Stein folgendermaßen: „Wenn es darum geht, Eigenschaften bestimmten Chromosomen zuzuordnen, muss man genetische Kartierungen durchführen. Dazu bedarf es der Entwicklung molekularer Marker.“ Das alles sei im Weizen sehr viel aufwendiger als in der Gerste. Denn für jedes Gen gibt es im hexaploiden Weizen in den meisten Fällen drei Kopien auf den drei homologen Genomen. „Die drei Teilgenome im Weizen sind sich auch nach vielen Millionen Jahren noch immer sehr ähnlich. Bei der Entwicklung molekularer Marker in Weizen, ist es oftmals sehr schwierig zu bestimmen, aus welchem der drei Weizengenome dieser stammt“, so Dr. Stein.
Der Startschuss
Das internationale Sequenzierkonsortium für Gerste wurde 2006 initiiert. Deutschland förderte das Projekt über das Bundesministerium für Bildung und Forschung (BMBF) in den letzten vier Jahren mit knapp sieben Millionen Euro. Zu Beginn des Vorhabens fassten alle Beteiligten den Entschluss, eine physikalische Karte des Gerstengenoms als Basis für die Entwicklung einer Referenzsequenz des Gerstengenoms zu erstellen. Anhand einer solchen Karte kann das Genom stückweise durch die Analyse sich überlappender BAC-Klone sequenziert und zu einer hoch-qualitativen Sequenz zusammengesetzt werden. Alternative Strategien wie die Shotgun-Sequenziertechnik können diesen Ansatz ergänzen, stellen aber gegenwärtig für komplexe Pflanzengenome noch keine vollwertige Alternative dar.
Erfahren Sie, warum die Erforschung des Erbguts der Gerste für Bier entscheidend ist und warum die genetischen Ressourcen der unterschiedlichsten Gerstensorten in Zeiten des Klimawandels an Bedeutung gewinnen. (Quelle: Climate Desk/youtube.com)
Vorarbeiten am kleinsten Gerstechromosom
In einer Pilotstudie prüfte das Wissenschaftler-Team um Dr. Stein zuerst, ob eine zumindest oberflächliche Sequenzierung einzelner Chromosomen möglich sei. Dazu wurde zunächst, im Labor des tschechischen Kooperationspartners Dr. Jaroslav Dolezel, Olomouc, das kleinste Gerstechromosom mit Hilfe eines Durchflußzytometers aus Chromosomensuspensionen gewonnen. Diese hatten die Wissenschaftler aus Wurzelmeristemen von Gerstenkeimlingen hergestellt. „Eine solche Auftrennung der Chromosomen direkt aus Gerste funktioniert nur für das kleinste, das 1H Chromosom. Die übrigen sechs Chromosomen sind sich in ihrer Größe zu ähnlich, als dass eine saubere Trennung per Durchflußzytometer möglich wäre“, so Dr. Stein.
Die chromosomale DNA von 1H wurde im Anschluss enzymatisch vervielfältigt, so dass den Wissenschaftlern genügend Ausgangsmaterial für weitere Arbeiten zur Verfügung stand. Zur Sequenzierung benutzten Dr. Stein und seine Kollegen eine sogenannte Next-Generation-Sequencing (NGS) Plattform von Roche/454. „Das Gerstengenom ist vollgestopft mit sich wiederholender, sog. repetitiver DNA. Daher mussten wir zunächst klären, welche Sequenzbereiche überhaupt zu kodierenden Genen gehörten“, erinnert sich Dr. Stein. Während der langwierigen Auswertung der umfangreichen Datensätze wurden die Wissenschaftler förmlich vom Fortschritt der Technik überholt. „Mit alternativen NGS-Plattformen könnten wir heute für deutlich weniger Geld eine viel höhere Abdeckung erzielen, als wir es vor zwei Jahren mit dem 454-Gerät konnten“, so Dr. Stein.
Bestimmung der Genanordnung
In den öffentlichen Datenbanken waren Mitte des letzten Jahrzehnts bereits über 500 Tausend kodierende Sequenzen für Gerste vorhanden. Diese können aus jedem Organismus unkompliziert über die Isolierung von Messenger RNA (mRNA) und die anschließende Sequenzierung von cDNA erstellt werden. Wo im Genom diese Sequenzen liegen, bleibt jedoch unbekannt. „Mit diesen Sequenzdaten kann man aber Marker entwickeln, mit denen man die Position eines Gens auf einem Chromosom bestimmen kann“, so Dr. Stein.

Blüten von Gerste im Vergleich. Gerste ist wie Weizen ein Selbstbefruchter. Bei der Selbstbestäubung gelangt der eigene Pollen auf die Narbe und befruchtet so die Eizelle der Blüte.
Bildquelle: © IPK Gatersleben
Anhand von Rekombinationsereignissen hatten Gerstenforscher bereits die relative Position von 3000 Gerstengenen im gesamten Gerstengenom bestimmt. Die etwa 500 Gene davon, die auf Chromosom 1H lagen, waren nun für Dr. Stein und seine Kollegen besonders interessant. Daran überprüften sie zunächst die Reinheit ihrer Chromosomenaufreinigung. Nur wenn der Großteil der von ihnen identifizierten codierenden Sequenzen tatsächlich mit den bereits bekannten 500 Sequenzen von Chromosom 1H übereinstimmte, wäre die Auftrennung der Chromosomen erfolgreich gewesen. „Zu 90 Prozent haben wir bei dieser Pilotstudie Gene von Chromosom 1H identifiziert. Das war ein gutes Ergebnis“, erinnert sich Dr. Stein und fährt fort: „Anhand der Sequenziertiefe wussten wir auch, dass wir das Chromosom rechnerisch maximal einfach repräsentiert hatten.“ Statistische Abschätzungen verrieten den Forschen, dass bei einer solchen Sequenziertiefe etwa 80 Prozent aller Gene erfasst werden. „Das war eine wichtige Information, denn wir wussten, dass wir zwar sehr viele Gene erfasst hatten, aber dass eben auch nicht wenige fehlten“, so Dr. Stein.
Die restlichen Gersten-Chromosomen
Nachdem die Vorgehensweise der Ansequenzierung einzelner Gerstechromosomen sehr vielversprechend verlaufen war, machten sich die Wissenschaftler an die übrigen Gerstenchromosmen. Diese konnten jedoch aufgrund ihrer ähnlichen Größe nicht direkt aus Gerste gewonnen werden. Eine andere Trennungsstrategie musste gefunden werden: Genetikern war Mitte des vergangenen Jahrhunderts im Rahmen zwischenartlicher Kreuzungen zwischen Gerste und Weizen gelungen, einzelne Gerstechromosomen in den Hintergrund des Weizengenoms einzukreuzen. Es entstanden sogenannte monosome Gerste-Weizen-Additionslinien. Für jedes Gerstechromosom, mit Ausnahme des kleinsten 1H, konnte eine Gerste-Weizenadditionslinie gewonnen werden. In diesen Additionslinien führten Genetiker dann Brüche in den addierten Gerstechromosomen herbei, woraus schließlich für jeden Gerstenchromosomarm eine sog. ditelosome Additionslinie in Weizen entwickelt werden konnte. Der jeweilige einzelne Gerstenchromosomenarm unterscheidet sich maßgeblich in der Größe von allen Weizenchromosomen, sodass er leicht mittels Durchflußzytometrie abgetrennt werden kann. Dies lieferte die Grundlage für die Ansequenzierung aller Gerstenchromosomen.

Der reife Fruchtstand der Gerste ist eine begrannte Ähre.
Bildquelle: © IPK Gatersleben
Analog zu der Pilotstudie konnten so alle Gerstechromosomen mit der Roche/454 Titanium Sequenziertechnik ansequenziert werden. „Wir haben uns gefragt, welche der identifizierten Gene bereits kartierten Markern bzw. Genen in anderen Gräsergenomen entsprechen“, erinnert sich Dr. Stein. Auf diese Weise überprüften die Forscher die Reinheit ihrer Daten. Aus vergleichenden Kartierungsstudien zwischen Gerste und Reis oder anderen bereits sequenzierten Gräsern war bekannt, welche Bereiche der Genome homolog waren. „Wir wussten beispielsweise, dass auf Reischromosom 1 und Gerstechromosom 3H, vereinfacht gesagt, im wesentlichen homologe Gene in konservierter Reihenfolge (orthologe Gene, collineare Genomabschnitte) vorkommen. So konnten wir jedem Bereich eines Gerstenchromosoms einen collinearen Bereich im Reischromosom zuordnen“, erklärt Dr. Stein. Diese Vergleiche vollzogen die Wissenschaftler um Dr. Stein auch mit dem Hirse (Sorghum bicolor) Genom und dem Genom des Wildgrases Zwenke (Brachypodium distachyon). Über den Vergleich konservierter Regionen konnten sie die Qualität ihrer Arbeit einschätzen.
Parallel zu den Sequenzierarbeiten in Deutschland nutzte eine schottische Arbeitsgruppe um Prof. Robbie Waugh die sortierten Chromosomen zur Hybridisierung auf einem Gerste-Microarray, der ca. 20.000 Gerstegene repräsentierte. Ein Teil der Gene konnte mit diesem Ansatz analog zur Chromosomensequenzierung eindeutig einzelnen Chromosomenarmen zugeordnet werden.
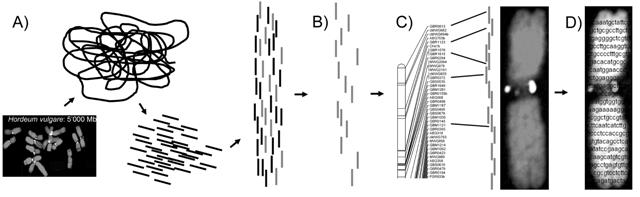
Der lange Weg von der DNA über die vollständige Genomsequenz bis zur physikalischen Genkarte. Eine Genkarte zeigt die lineare Anordnung der Gene im Genom. Auf einer physikalischen Karte werden neben der Reihenfolge auch die genauen Abstände zwischen den Genen eingetragen.
Bildquelle: © IPK Gatersleben
Mittels der beiden komplementären Ansätze, Sequenzierung und Array-Hybridisierung, konnten Dr. Stein und Kollegen innerhalb von zwei Jahren somit ein Modell der linearen Abfolge von über zwei Drittel aller Gerstengene erschaffen. „Wir haben die bisher genauste Annäherung an das Gerstengenom geschaffen, die im Moment die beste Grundlage für molekulargenetische Analysen bietet“, fasst Dr. Stein die gemeinsamen Arbeiten zusammen. „Unser Modell besitzt zu 90 Prozent Vorhersagekraft. Wenn ein Forscher also ein Gen auswählt, wird er dieses auch mit 90 prozentiger Wahrscheinlichkeit an dem von uns bestimmten Ort finden“, so Dr. Stein. Genomweite Polymorphismusanalysen sind nun mit diesen Sequenzinformationen auch im Gerstengenom möglich. Diese sind die Grundlage für die moderne, genombasierte Züchtungsforschung). Eine sogenannte Referenzsequenz für Gerste haben Dr. Stein und seine Kollegen allerdings mit diesen Arbeiten noch nicht erstellt. Dafür reichte die Sequenziertiefe nicht aus.
Die Arbeiten zur Ansequenzierung sortierter Gerstechromosomen macht bereits Schule: Das internationale Weizensequenzierkonsortium (IWGSC) übernimmt die erfolgreiche Vorgehensweise von Dr. Stein und seinen Kollegen exakt. Gegenwärtig werden alle Weizenchromosomen auf Basis sortierter Chromosomenarme ansequenziert. Lediglich bei der Sequenzierung profitiert das Weizen-Konsortium von der fortschrittlicheren Illumina Technologie.
Stand der Dinge
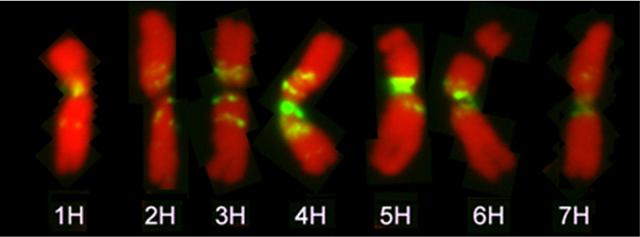
Mit Hilfe einer Fluoreszenz-Hybridisierung markierter Karyotyp der Gerste, am Beispiel von Chromosomen in der Metaphase bei der Mitose der Zellteilung.
Bildquelle: © IPK Gatersleben
Die physikalische Karte des Gerstengenoms, die zur kartengestützten, vollständigen Sequenzierung des Gerstengenoms benötigt wird, kann bis Herbst diesen Jahres von Forscherteams aus Europa, den USA, Japan und Australien unter Leitung des IPK und im Rahmen des „International Barley Sequencing Consortium“ (IBSC), fertig gestellt werden. Am IPK in Gatersleben hat außerdem bereits auf Basis dieser physikalischen Karte die Sequenzierung des Gerstenchromosoms 3H begonnen. Am französischen Institut National de la Recherche Agronomique (INRA) arbeiten Wissenschaftler parallel an der Sequenzierung des sehr ähnlichen Weizenchromosoms 3B. In einer Kooperation zwischen beiden Instituten steht die Analyse der Gemeinsamkeiten und Unterschiede zwischen diesen verwandten Chromosomen aus Gerste und Weizen im Zentrum des Interesses.
Auf die vollständige Sequenz des Gerstegenoms werden Wissenschaftler und Züchter laut Dr. Stein nicht mehr lange warten müssen: „Mit dem festen Vertrauen darauf, dass wir weitere Innovationen im Bereich der Sequenziertechnologie erwarten können, sollte eine hochqualitative Referenzsequenz für Gerste innerhalb der nächsten zwei Jahre realistisch erreichbar sein.“
Quelle:
Mayer, K. F.X. et al. (2011): Unlocking the Barley Genome by Chromosomal and Comparative Genomics. In: The Plant Cell, (April 2011), doi:10.1105/tpc.110.082537.
Zum Weiterlesen:
- Herausforderungen bei der Genomsequenzierung von Pflanzen
- Erster Schritt zur Erbgut-Entschlüsselung von Getreide
- 517 Reissorten in Stücke zerlegt
- An Arabidopsis führt kein Weg vorbei
- Von der Wildpflanze zur Kulturpflanze
Einige Pflanzensteckbriefe:
