


Regulation der Genaktivitäten besser verstehen
Prognosemodelle für Transkriptionsfaktoren werden zunehmend genauer

Modelle: Selbstlernende Algorithmen eröffnen neue Ansätze in der Molekularbiologie. (Bildequelle: © iStock.com/ktsimage)
Ob ein Gen aktiviert wird oder nicht, hängt von Transkriptionsfaktoren ab. Welche Transkriptionsfaktoren mit welchen DNA-Bereichen interagieren, versuchen Molekularbiologen mit Modellen vorherzusagen. Neue Ansätze machen diese immer verlässlicher.
Die Gene bestimmen, welche Proteine eine Zelle grundsätzlich herstellen kann. Doch ob – und unter welchen Umständen – das tatsächlich geschieht, hängt fast immer von Transkriptionsfaktoren ab. Dabei handelt es sich um Proteine, die bestimmte Sequenzen der DNA erkennen und dort beispielsweise weitere, für die Transkription wichtige Proteine binden. Oder sie verändern die Struktur des Chromatins, um eine Transkription zu ermöglichen.
Um vorherzusagen, in welchen Zelltypen und unter welchen Bedingungen ein Gen tatsächlich seine Aktivität entfaltet, ist ein präzises Verständnis über die Interaktion von Transkriptionsfaktoren mit der DNA wichtig. Die Molekularbiologie hat dazu eine Vielzahl von Modellen und Datenbanken entwickelt, um letztlich vorhersagen zu können, wie sich eine bestimmte Veränderung des Erbguts auf die Genaktivität auswirkt. Präzise Prognosewerkzeuge zur Aktivität von Transkriptionsfaktoren könnten so auch die Pflanzenzüchtungsforschung deutlich beschleunigen.
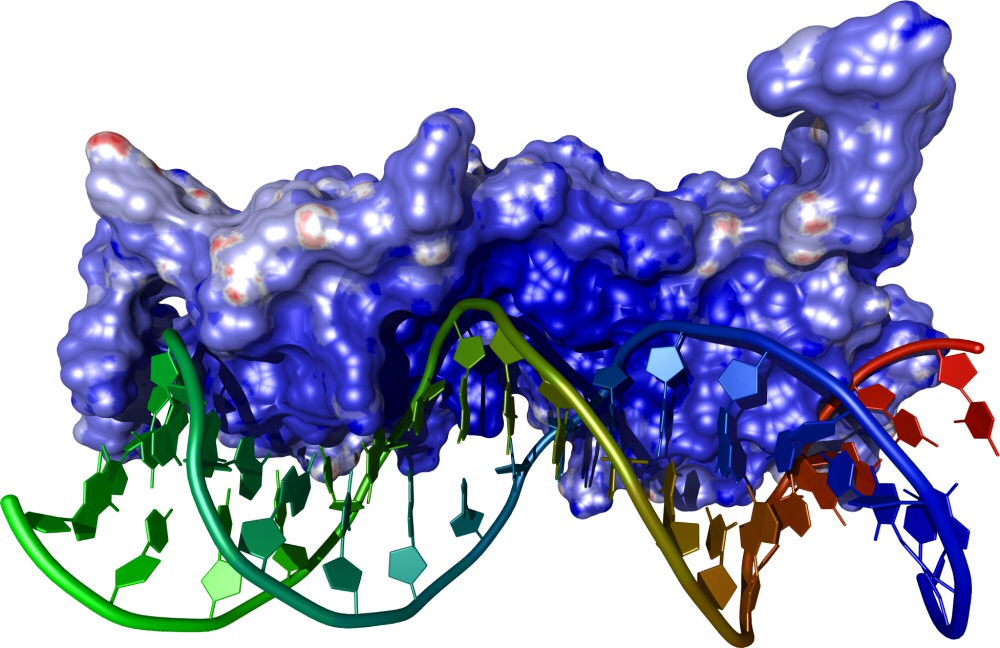
Darstellung eines Transkriptionsfaktors, der sich an die passende DNA-Sequenz legt.
Bildquelle: © Anmoll/Wikimedia.org; CC-BY-SA-2.0-DE
Bestehende Vorhersagemodelle haben Schwächen
Die vorhandenen Modelle haben jedoch nur beschränkte Einsatzfelder und kämpfen mit methodischen Schwächen oder unvollständigen Datengrundlagen. Biologen der Humboldt-Universität Berlin und der Universität Grenoble haben nun eine umfassende Übersicht der existierenden Modelle mit ihren Stärken und Schwächen zusammengestellt und dabei auch aufgezeigt, wie künftige Modelle deutlich leistungsfähiger werden könnten.
Wesentlicher Anteil kommt dabei zunächst der Identifikation jener Stellen zu, an die die jeweiligen Transkriptionsfaktoren binden. Dabei berücksichtigen die Forscher neben der DNA-Sequenz deren Position im Nukleosom, den Chromatinstatus, Methylierungsmuster, die 3D-Struktur des Genoms und ein mögliches kooperatives Bindungsverhalten mehrerer Transkriptionsfaktoren. Nicht zuletzt ist auch die quantitative Modellierung des Bindungsvorgangs entscheidend, um Genaktivitäten zu prognostizieren. „Es wurden große Fortschritte dabei gemacht, Transkriptionsfaktoren zu kartieren, aber die resultierenden Modelle sagen die tatsächliche Genregulation oft nur schlecht voraus“, stellen die Autoren in ihrer Studie fest.
Drei Arten von Bindungseinflüssen berücksichtigen
Modelle müssen drei unterschiedliche Mechanismen berücksichtigen, wie Transkriptionsfaktoren ihre Bindungsstellen erkennen: (1) die physikalische Interaktion zwischen den Aminosäureseitenketten und den Rändern der komplementären DNA-Basen infolge von Wasserstoffbrücken, hydrophoben Wechselwirkungen und Salzbrücken; (2) die Interaktion zwischen dem Transkriptionsfaktor und dem Phosphatrückgrat der DNA; und (3) strukturelle Eigenschaften der DNA wie deren Krümmung, Wicklung und der Größe ihrer Furche.
Der bisherige Goldstandard, um genomweit Bindungsstellen für einen bestimmten Transkriptionsfaktor in vivo zu entdecken, ist die ChIP-seq (Chromatin ImmunoPrecipitation DNA-Sequencing), eine Kombination aus Chromatin-Immunpräzipitation und DNA-Sequenzierung im Hochdurchsatz. Varianten dieser Methode haben inzwischen einige ihrer ursprünglichen Schwächen wie eine hohe Rate falsch-positiver Treffer verloren. Eine noch junge Ergänzung dazu bietet die In-vitro-Methode DAP-seq (DNA Affinity Purification Sequencing), die fragmentierte genomische DNA als Substrate für die Immunopräzipitation sowie rekombinante Transkriptionsfaktoren nutzt. Auch sie hat Schwächen, da manche Transkriptionsfaktoren rekombinant nicht stabil sind oder zusätzliche Bindungspartner benötigen. Die Kombination beider Methoden führe allerdings zu guten Vorhersagen, so das Fazit der Studie.
Übersicht: Nukleobasen, auch Nukleinbasen
Strukturinformationen komplementieren Sequenzinformationen
Um die Genauigkeit weiter zu verbessern, müsse man zusätzlich komplexe Eigenschaften wie Abhängigkeiten von der Sequenzposition innerhalb des DNA-Strangs und der DNA-Form berücksichtigen, erläutern die Forscher. Einen guten Ansatz nach diesem Prinzip verfolge beispielsweise das Programm Morpheus. Es ermöglicht Bindungsstellenprognosen auf Basis der etablierten Positions-Gewichts-Matrix, die die Wahrscheinlichkeit bestimmter Sequenzfolgen beschreibt, ergänzt diese aber um den Einfluss der Sequenzposition. Damit seien bereits verbesserte Vorhersagen für pflanzliche Transkriptionsfaktoren gelungen.
Schwächen bisheriger Modelle bei der bisherigen Berücksichtigung von Formparametern erklären die Forscher damit, dass zwar das Kernmotiv der Bindungsstellen der Haupteinflussfaktor sei, aber eben nicht der alleinige. Angrenzende DNA-Regionen haben ebenfalls Einfluss, doch über sie liegen oft zu wenige Sequenzinformationen vor. Jüngere Modelle beziehen allerdings bereits 13 Form-Parameter ein. Ihre Prognosen übertreffen jene rein sequenzbasierte Modelle, beruhen aber auf computergenerierten Simulationen der 3D-Strukturen. Eine weitere Verbesserung wäre hier die Verwendung röntgenkristallographischer experimenteller Strukturdaten, so die Empfehlung der Forscher.
Selbstlernende Algorithmen eröffnen neue Ansätze
Ein weiterer interessanter Ansatz seien Methoden unter Zuhilfenahme künstlicher Intelligenz. Selbstlernende Algorithmen brächten nicht nur enorme Zeitersparnisse. Sie kämen auch mit Signalrauschen besser zurecht und könnten mit vielen unterschiedlichen Formen an Rohdaten trainiert werden. Beispiele für erfolgreiche Algorithmen sind DeepBind, DeepSEA, TFImpute, DeFind und DFIM.
Trotz der Vielfalt an Modellen und deren kontinuierlicher Verbesserung gebe es noch kein Allzweck-Modell, das für alle Transkriptionsfaktoren die optimalen Ergebnisse liefert, resümieren die Forscher. Erfolgversprechend sei in jedem Fall, die „Breite“ des Next-Generation-Sequencings mit der „Tiefe“ der Struktur-Ansätze zu kombinieren.
Quelle:
Lai, X. et al. (2019): Building Transcription Factor Binding Site Models to Understand Gene Regulation in Plants. In: Molecular Plant 12, 743–763, (Juni 2019), doi: 10.1016/j.molp.2018.10.010.
Zum Weiterlesen auf Pflanzenforschung.de:
- Zusammenspiel der Transkriptionsfaktoren - Wie Pflanzen das Ablesen ihrer Gene steuern
- Molekulare Symphonien - Transkriptionsfaktoren exprimieren tausende Gene
- Tool mit neuen Möglichkeiten: MapMan4 - So lassen sich Proteinfunktionen schneller identifizieren
Titelbild: Modelle: Selbstlernende Algorithmen eröffnen neue Ansätze in der Molekularbiologie. (Bildequelle: © iStock.com/ktsimage)
