


Die Entschlüsselung der Süßen

Samenträger nach beendeter Blüte. (Quelle: © KWS Saat AG)
Mehr als 250 Jahre nachdem das wirtschaftliche und wissenschaftliche Interesse an der Rübe durch den Nachweis von Zucker in der Runkelrübe geweckt wurde, erstellen Wissenschaftler im GABI-Future-Projekt eine Referenzsequenz des Genoms der Zuckerrübe.
Daniela Holtgräwe, Bernd Weisshaar, Heinz Himmelbauer
Mehr als 250 Jahre nachdem das wirtschaftliche und wissenschaftliche Interesse an der Rübe durch den Nachweis von Zucker in der Runkelrübe geweckt wurde, entschlüsseln Forscher vom Max-Planck-Institut für molekulare Genetik in Berlin und dem Centrum für Biotechnologie (CeBiTec) der Universität Bielefeld unter Beteiligung deutscher Saatgutfirmen die gesamte Erbinformation der Zuckerrübe (Beta vulgaris ssp. vulgaris). Das Projekt BEETSEQ wird im Rahmen der Förderinitiative GABI-FUTURE vom Bundesministerium für Bildung und Forschung gefördert.
Die Historie und Gegenwart der Süßen
Die Zuckerrübe gehört zur Gattung Beta, deren Stammform ihren Ursprung vermutlich an der Mittelmeer- und Nordseeküste hat. Der deutsche Chemiker Andreas Sigismund Marggraf dokumentierte 1747 den Zuckergehalt der Runkelrübe und entdeckte ferner, dass der Zucker aus der Rübe mit dem aus Zuckerrohr identisch ist. Durch Züchtung und Selektion auf einen hohen Zuckergehalt entstand aus der Runkelrübe die Zuckerrübe. Mit der Zeit konnte der Zuckergehalt im weißen Wurzelkörper von ehemals ca. 3-5% immer weiter auf einen Gehalt von ca. 18-20% in modernen Zuckerrüben gesteigert werden. Derzeit stammt weltweit etwa ein Drittel des Zuckers aus Zuckerrüben, sodass die Zuckerrübe heute neben dem Zuckerrohr die wichtigste Zuckerpflanze darstellt.
Zuckerrüben werden auf den besten Böden in den gemäßigten Breiten, hauptsächlich in Europa, den USA und in Russland kultiviert. Für einen hohen Ertrag benötigt sie gemäßigte Temperaturen, viel Licht und viel Wasser sowie nährstoffreiche Böden. Die Zuckerrübe ist eine zweijährige, fremdbefruchtende und diploide Nutzpflanze. Im ersten Jahr bildet sie eine Blattrosette und den Rübenkörper aus. Nach einer Vernalisationsperiode (die natürliche Kältebehandlung der Rüben im Winter) geht sie erst im zweiten Jahr in die generative Phase über und bildet Blütenschosse und Samenträger aus. Die derzeitigen Hauptproduzenten von Zucker aus Zuckerrüben sind Deutschland, Frankreich und Polen. EU-weit werden jährlich etwa 120 Mio. Tonnen Zuckerrüben produziert, aus denen ca. 14 – 16 Mio. Tonnen Kristallzucker gewonnen werden. Der Zucker kommt entweder direkt in den Handel oder wird in der Lebensmittelindustrie als Süßmittel eingesetzt.
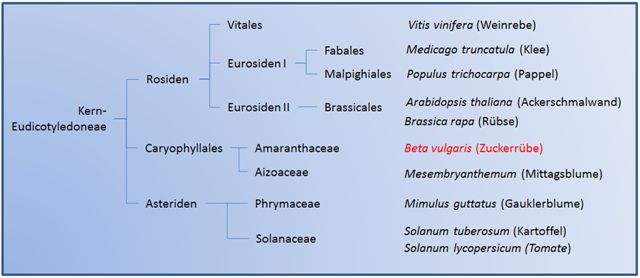
Einordnung der Zuckerrübe in Relation zu ausgewählten Eudikotyledonen in einen vereinfachten Stammbaum (verändert nach Dohm et al., 2009; Quelle: GenomXpress 2/2011, S. 5).
In den letzten 15 Jahren verfünffachte die chemisch-technische Industrie in Deutschland ihren Verbrauch an Zucker hauptsächlich durch den Zuwachs im biotechnologischen Produktionssektor. Außerhalb des Ernährungsbereichs wird der Zucker zur Produktion von biologisch abbaubaren Kunststoffen für die Herstellung von Bechern, Folien, Polsterungen, Klebstoffen, Kosmetika und Lacken verwendet, oder als Nahrungsgrundlage für Bakterien und Pilze eingesetzt, welche hieraus Organische Säuren, Antibiotika und Vitamine produzieren. Jüngst gewinnen Zuckerrüben als nachwachsender Rohstoff, z.B. zur Produktion von Bioethanol und Biogas, an ökonomischer Bedeutung.
Die Sequenzierungsstrategie
Das zentrale Ziel des Beetseq-Projekts ist die Erstellung einer genomweiten Referenzsequenz der Zuckerrübe basierend auf dem doppelt haploiden und damit komplett homozygoten (reinerbigen) Genotyp KWs2320. Die Zuckerrübe hat einen Chromosomensatz von 2n = 18 Chromosomen. Das haploide Genom der Zuckerrübe ist nach Untersuchungen von Arumuganathan und Earle (1991) etwa 758 mbp groß. Damit ist es fünfmal größer als das von Ackerschmalwand (Arabidopsis thaliana), aber nur etwa halb so groß wie das Rapsgenom. Da die Größe eines Genoms mit der Genomkomplexität und dem Organisationsgrad des Organismus in keinem eindeutigen Zusammenhang steht, ist es nicht allzu verwunderlich, dass das Zuckerrübengenom zwar nur etwa ein Viertel so groß wie das menschliche Genom ist, aber vermutlich mehr Gene als das Humangenom enthält. Ferner verfügt die Zuckerrübe, wie viele komplexe Genome von Eukaryonten, über einen erheblichen Anteil sich nahezu identisch wiederholender „repetitiver“ Sequenzen.
Um eine hochwertige Referenzsequenz verfügbar machen zu können, werden bei der „de novo“-Sequenzierung der Zuckerrübe verschiedene Sequenzierungstechnologien und -strategien mit den Positionsinformationen einer physikalischen BAC-Klonkarte kombiniert. Die Erstellung der Daten für die Genomassemblierung wird unter Anwendung neuer Sequenziertechnologien und traditioneller Sanger-Sequenzierung bewerkstelligt. Insbesondere werden relativ kostengünstige „Whole Genome Shotgun“ (WGs) Datensätze und Daten aus „Paired-End“-Sequenzierung kombiniert. Während für die WGs-Sequenzierung mit „single reads“ ausschließlich auf die 454-Technologie gesetzt wird, kommen bei der „Paired-end“-Sequenzierung sowohl die 454-Technologie als auch die Solexa-Technologie zum Einsatz.
Diese paarigen Sequenzen, generiert aus den Enden fragmentierter DNA-Moleküle definierter Länge zwischen 1 kbp und 20 kbp, können aufgrund ihrer Abstandsinformation zur Überbrückung von repetitiven Bereichen bei der Assemblierung genutzt werden. Größere Distanzen oder Sequenzlücken können über mittels Sanger-Technologie generierten paarigen Endsequenzen von genomischen BAC- und Fosmidklonen überspannt werden. Die korrekte Assemblierung und Integration dieser sehr unterschiedlichen Typen von Sequenzdatensätzen, sowie die Berücksichtigung von Positionsinformationen aus genetischen und physikalischen Karten bei der Verrechnung stellen eine große Herausforderung für die Bioinformatik dar.
An der Fertigstellung der Referenzsequenz wird im Rahmen des Beetseq-Projektes weiter intensiv gearbeitet. Als Grundlage für die Ermittlung der Gendichte, für Genvorhersagen und Genannotierung, werden Transkripte aus unterschiedlichen Geweben sequenziert. Neben der Erstellung einer Referenzsequenz basierend auf dem zentralen Genotyp KWs2320, werden für weitere doppelhaploide Genotypen der Zuckerrübe, sowie für Genome von verschiedenen Vertretern der Gattung Beta und auch von nah verwandten Arten, beispielsweise von Spinat, Genomassemblies errechnet.

Trennwände in einem Zuckerrüben-Züchtungsprogramm.
Bildquelle: © KWS SAAT AG
Genomsequenz mit Relevanz für Wissenschaft und Wirtschaft
Die in der Referenzsequenz enthaltene detaillierte Information über die molekulare Organisation des Erbmaterials der Zuckerrübe wird, unterstützt durch die genomischen Sequenzdatensätze weiterer Genotypen und verwandter Arten, die Forschung im Bereich der vergleichenden Genomanalyse vorantreiben. Die molekulare Identifizierung von artspezifischen Genen oder Genstrukturen ist wissenschaftlich von großem Interesse. In den vergangenen Jahren sind für einige Vertreter der Blütenpflanzen vollständige Genomsequenzen veröffentlicht worden, darunter Ackerschmalwand, Weinrebe, Pappel, Reis, Papaya, Mais, Apfel und Sojabohne.
Die Genomsequenzierung vorwiegend wirtschaftlich bedeutender Arten wird weiterhin international vorangetrieben. Die Zuckerrübe ist mit keiner der hier genannten Arten eng verwandt, sodass das Gebiet der vergleichenden Genomanalyse eine interessante neue Forschungsgrundlage erhält. Neben den übergreifenden Fragestellungen zur Pflanzenevolution kann die Analyse der genomischen Sequenz der Zuckerrübe auch Fragestellungen bezüglich einzelner Gene oder Genfamilien beantworten helfen, oder Hinweise für die Entwicklung spezifischer Merkmale, wie beispielsweise die Ausbildung des zuckerhaltigen Rübenkörpers oder die Zweijährigkeit geben. Nicht zuletzt wird der modernen Züchtungsforschung basierend auf der Grundlage der Genomsequenz die Entwicklung molekularer Marker enorm erleichtert und somit die Züchtung leistungsfähigerer Zuckerrübensorten vereinfacht.
Dieser Artikel wurde Pflanzenforschung.de im Rahmen einer Kooperation mit dem Forschungsmagazin GenomXpress zur Verfügung gestellt. Der GenomXpress ist hier downloadbar.
Quellen:
- C. Lange et al. (2008): Construction and characterization of a sugar beet (Beta vulgaris) fosmid library. Genome 51(11): 948–951.
- Dohm JC, et al. (2009: Haplotype divergence in Beta vulgaris and microsynteny with sequenced plant genomes. Plant J., Jan;57(1):14-26.
- Lange C, et al. (2010): Highthroughput identification of genetic markers using representational oligonucleotide microarray analysis. Theor Appl Genet. Aug;121(3):549-65.
- Dohm JC, et al. (2011): A comprehensive gene-based physical BAC map of sugar beet (Beta vulgaris) by integration of physical and genetic mapping. Zur Veröffentlichung eingereicht.
