


Neue Methode zur Strukturaufklärung von RNA
Tiefensequenzierung und räumliche Nähe von Atomen für 3D
Wie sich ein RNA-Molekül räumlich ausrichtet, ist wichtig für seine Funktion, aber nicht immer einfach zu klären. Eine neue Methode verspricht Abhilfe. (Bildquelle: © iStock.com/JVisentin)
RNAs sind an zahlreichen zellulären Prozessen beteiligt. Doch wie diese kleinen Moleküle in Natura aussehen, lässt sich oftmals nur erahnen, denn die Strukturaufklärung von RNA-Molekülen ist bisher sehr aufwendig und manchmal auch gar nicht möglich. Eine neue Methode verspricht Abhilfe.
Genau wie die DNA besteht auch die RNA (ribonucleic acid) aus einem Rückgrat aus Zucker-Phosphat und einer Abfolge von vier verschiedenen Basen. Im Unterschied zur DNA ist der Zucker in der RNA die Ribose, und die Base Thymin ist ersetzt durch Uracil. Und im Gegensatz zur DNA liegt die RNA nicht als Doppelhelix, sondern als einzelner Strang vor. Ihre Aufgaben innerhalb eines Organismus sind essentiell und äußerst vielfältig und reichen von der Umsetzung von Erbgutinformationen bis zur Genregulation.
3D- Struktur bestimmt die Funktion
RNA kommt in verschiedenen Varianten vor:
Die Boten-RNA (mRNA, Messenger-RNA) transportiert die genetische Information aus dem Zellkern zu den Ribosomen, wo die Proteine dem Bauplan der DNA entsprechend hergestellt werden. Daran ist die Transfer-RNA (tRNA) beteiligt. Sie steuert in den Ribosomen den Einbau einzelner Aminosäuren in die wachsende Proteinkette. An der Reifung der mRNA sind die snRNA (small nuclear RNA) und die snoRNA (small nucleolar RNA) beteiligt.
Die ribosomale RNA (rRNA) trägt, ähnlich wie die tRNA, keine genetische Information, sondern ist am Aufbau des Ribosoms beteiligt. Die siRNAs, small interfering RNA, entsteht bei der sogenannten RNAi (RNA-Interferenz). Eng verwandt mit den siRNAs sind die micro RNAs (miRNAs): Beide erfüllen essentielle Funktionen bei der Regulation von zellulären Prozessen.
Einzelsträngige RNAs kommen in der Natur nicht immer als lineares Molekül vor. Im Gegenteil: Viele RNAs falten sich zu komplexen sekundären und tertiären Strukturen, die von den Wasserstoff-Bindungen zwischen den Nukleobasen abhängen. Ohne diese räumliche Anordnung wären viele der katalytischen, regulatorischen und informationstragenden Eigenschaften der RNA-Moleküle gar nicht vorhanden. Für Wissenschaftler sind solche Strukturen nicht immer einfach vorhersehbar und verleihen den RNA-Molekülen eine gewisse Unberechenbarkeit, vor allem dann, wenn sie zu wissenschaftlichen oder therapeutischen Zwecken synthetisch hergestellt werden.
12 verschiedene Familien
Daher ist es für Wissenschaftler essentiell, RNA-Moleküle zur klassifizieren: Alle Interaktionen zwischen Basen, die über mindestens zwei gewöhnliche Wasserstoff-Bindungen verfügen, können in 12 verschiedene Familien eingeteilt werden. Jede Familie besteht aus einer 4 x 4 Matrix der vier RNA-Basen: U, C, A, G. Die gewöhnlichen Watson-Crick-Paare gehören zu einer dieser Familien, alle anderen 11 Familien beinhalten keine Watson-Crick-Paare.
Die Watson-Crick-Paare bilden die doppelsträngige Haarnadelstruktur der RNA-Sekundär-Struktur aus. Die übrigen Familien bilden die sogenannten RNA-Module aus – die Bausteine der Tertiärstruktur. Sie sind auch an den intramolekularen Verbindungen beteiligt.
Erst die Sekundär-, dann die Tertiärstruktur
Wie sich die RNA in der Natur anordnet, hängt also in erster Linie von den Haarnadelstrukturen ab, die die Watson-Crick-Basen ausbilden. Aber auch die Nicht-Watson-Crick-Paare haben Einfluss auf die räumliche Anordnung und die Funktion von RNA-Molekülen. Viele computergestützte Ansätze zur Aufklärung der RNA-Struktur folgen genau diesem Ansatz: Zuerst wird die Sekundärstruktur ermittelt, dann die Tertiärstruktur, also die räumliche Anordnung des Moleküls.
Bisherige Strukturaufklärung von RNAs
Um die räumlichen Strukturen bestimmten Funktionen zuordnen zu können, ist es wichtig, diese aufzuklären. Bisher nutzte man dafür entweder die auf Röntgenstrahlung basierende Kristallographie, die nukleare Magnetresonanzspektroskopie oder auch die Elektronenmikroskopie. Doch alle Methoden sind sehr zeitaufwendig und benötigen teilweise große RNA-Mengen als Ausgangsmaterial. Zudem sind die optimalen Lösungs- und Kristallisationsbedingungen für jedes RNA-Molekül nicht immer bekannt und müssen mühsam ausprobiert werden.
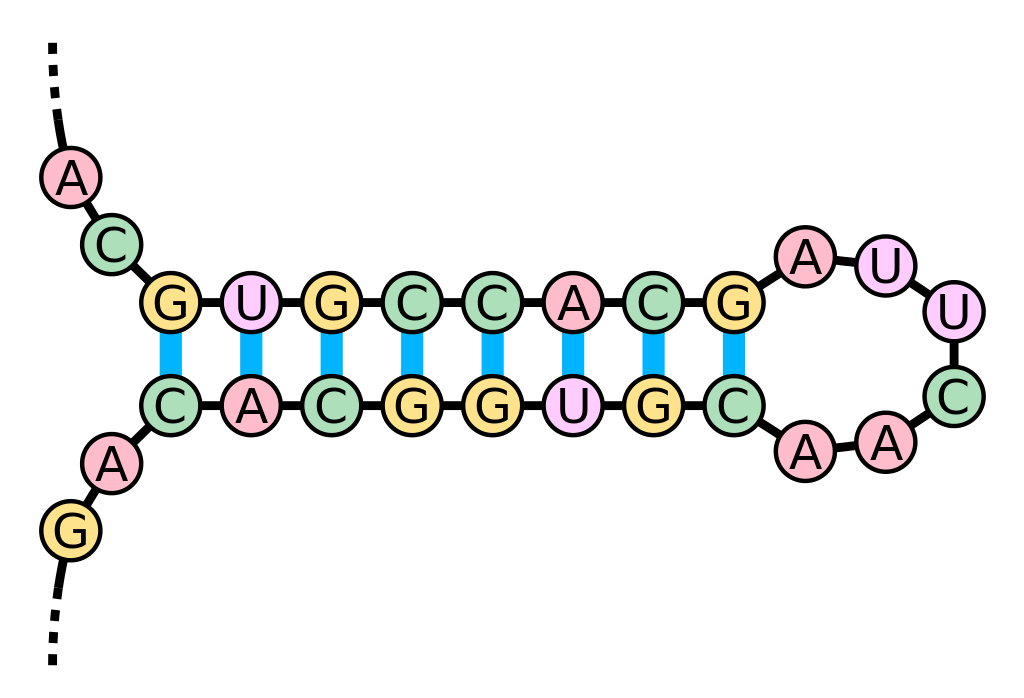
Beispiel für eine RNA-Haarnadelstruktur.
Bildquelle: © Sakurambo / wikimedia.org; CC BY-SA 3.0
Auch phylogenetische Ansätze können hilfreich sein, wenn es darum geht, die RNA-Struktur aufzuklären. Denn homologe Sequenzen bilden häufig ähnliche, räumliche Strukturen aus. Doch um auf diese Methode zugrückgreifen zu können, brauchen Wissenschaftler viele Daten zu Sequenzvariationen. Und die sind nicht immer vorhanden.
Andere in silico Ansätze beruhen auf mathematischen Modellen, die die RNA-Moleküle so anordnen, dass möglichst wenig freie Energie auftritt und möglichst viele Basen gebunden sind. Obwohl diese rein theoretischen Ansätze ständig verbessert werden, bleiben sie mathematisch komplex und können zu mehreren Anordnungsvarianten führen. Ihre Genauigkeit liegt derzeit erst bei 75 bis 80 Prozent.
Auch chemische und enzymatische Sonden können dabei helfen, die räumliche RNA-Struktur zu ergründen. Ausgesucht werden die Chemikalien je nach Reaktivität mit bestimmten Atomen der Basen oder des Zucker-Phosphat-Rückgrats; bei Enzymen kommt es darauf an, ob sie RNAs bevorzugt an gepaarten oder ungepaarten Regionen schneiden. Auch darüber lässt sich mit Hilfe von Computermodellen die RNA-Struktur vorhersagen.
Diese Ansätze wurden in den letzten Jahren auch vermehrt mit Tiefen-Sequenzierungsansätzen kombiniert. Doch gepaarte RNA-Stränge oder Nukleotide können hiermit nicht identifiziert werden. Hier müssen Wissenschaftler wieder auf phylogenetische Analysen und Computermodelle zurückgreifen.
Im komplexen RNA-Struktur-Puzzle müssen alle Informationen zur räumlichen Nähe und zur Ausbildung von Basenpaaren in einer ausreichend hohen Auflösung vorhanden sein. Erst dann können Wissenschaftler mit Hilfe von Computermodellen Rückschlüsse auf die RNA-Struktur ziehen.
Neue Methode zur Strukturaufklärung
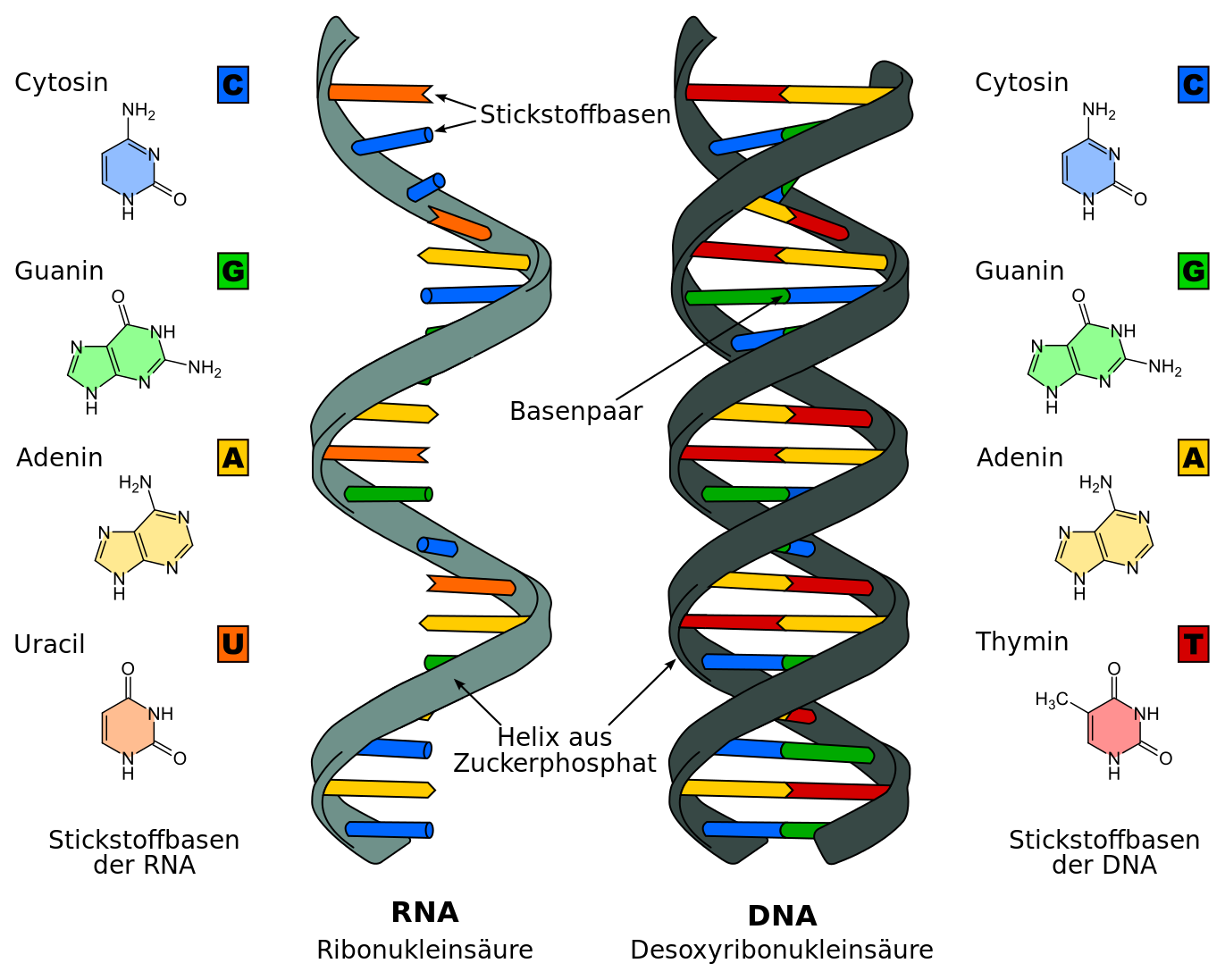
DNA und RNA im Vergleich: Genau wie die DNA (deoxyribonucleic acid, dt. Desoxyribonukleinsäure) besteht auch die RNA (ribonucleic acid, dt. Ribonukleinsäure) aus einem Rückgrat aus Zucker-Phosphat und einer Abfolge von vier verschiedenen Basen. Allerdings ist der Zucker in der RNA die Ribose und die Base Thymin ist ersetzt durch Uracil. Darüber hinaus liegt die RNA nicht als Doppelhelix, sondern als einzelner Strang vor.
Bildquelle: © Sponk / wikimedia.org; CC BY-SA 3.0
In einer aktuellen Studie stellen Wissenschaftler nun eine neue Methode vor, wie sich die räumliche Struktur von RNA wesentlich schneller aufklären lässt als bisher. Die Forscher nennen Ihre Methode zur dreidimensionalen Strukturaufklärung „RNA proximity ligation” (RPL), also die Ligation von räumlich benachbarter RNA. In ihrem Ansatz beschreiben die Wissenschaftler eine Tiefen-Sequenzierung und eine Ligation benachbarter Atome, mit der sie aus einem komplexen Mix aus nicht-denanturierten RNA-Transkripten aus Hefezellen ohne Zellwand oder aus menschlichen Zelllinien Informationen über die räumliche Nähe von Nukleotiden erhalten.
Zunächst lassen die Forscher RNasen (endogene RNasen bei den Hefezellen, exogene bei den humanen Zellen) die RNA schneiden. In einem nächsten Schritt geben sie dem Mix eine exogene T4 RNA Ligase zu, die nach dem Zufallsprinzip freie RNA-Enden miteinander verknüpft. Die so entstandenen Moleküle sequenzieren die Forscher. Die Sequenzierergebnisse werden dann mit der bekannten Primärstruktur - also der einfachen Basenabfolge des RNA-Moleküls - verglichen.
Die Experimente der Forscher zeigten, dass die Ligationen in den allermeisten Fällen intramolekular, also nicht zwischen verschiedenen RNA-Molekülen stattfinden. Doch die Ergebnisse sind keinesfalls klar und eindeutig, denn die Forscher entdeckten zufällige Ligationen, die ein großes Hintergrundrauschen in den Daten verursachten. Um das Hintergrundrauschen bei den Ligationen zu verbessern, analysierten die Forscher ihre Daten in Fenstern mit jeweils 21 Nukleotiden. So erfassen sie mehr Ligationen von Molekülenden, die sich in räumlicher Nähe zueinander befinden. Auf Basis dieser Ergebnisse erstellen die Forscher RPL-Scores, die wiederum in Karten mit möglichen räumlichen Strukturen der RNA umgewandelt werden.
Der große Vorteil der Methode: Sie eignet sich zum Hochdurchsatzverfahren und damit zur Automatisierung. Der Nachteil: Im Moment können nur 21-Basenpaar-Fenster analysiert werden, was durchaus noch verbesserungsbedürftig ist.
Nach den einfacher umsetzbaren Methoden zur DNA-Aufklärung folgt der nächste große Schritt, um die in einer Zelle ablaufenden Prozesse detaillierter aufzuklären. Egal ob für die Therapie von Krankheiten oder eine verbesserte Reaktion auf bestimmte Umwelteinflüsse, die Ribonukleinsäure (RNA) wird die Forschung, aber auch die Anwendung stark verändern. Erinnert sei in diesem Zusammenhang an bereits laufende Versuche, RNA-Moleküle großflächig zu nutzen, um Krankheiten und Schädlinge zu bekämpfen, ohne die Pflanzen gentechnisch zu verändern. Aber das ist nur eine Möglichkeit. Basis für alle ist das bessere Verständnis und dieses wird durch die hier vorgestellte Methode maßgeblich verändert.
Quelle:
Ramani, V. et al. (2015): High-throughput determination of RNA structure by proximity ligation. In: Nature Biotechnology; 33(9):980-4, (September 2015), doi: 10.1038/nbt.3289.
Weiterlesen auf Pflanzenforschung.de:
- Mikro-RNAs in Pflanzen: Neuer Regulator entdeckt
- Kleine RNAs regulieren die Abwehr in der Tomate
- Direkte RNA-Sequenzierung
- Pflanzliche RNA greift in den Stoffwechsel ein
- RNA-Viren-Abwehr - In allen höheren Lebewesen immer bereit
Titelbild: Wie sich ein RNA-Molekül räumlich ausrichtet, ist wichtig für seine Funktion, aber nicht immer einfach zu klären. Eine neue Methode verspricht Abhilfe. (Bildquelle: © iStock.com/JVisentin)
